Abstract
It has been previously shown [1] that S. cerevisiae proteins preferentially interact with proteins of the same estimated likely time of origin. Using a similar approach but focusing on a less broad evolutionary time span I observed that the likelihood for protein interactions depends on the proteins’ age. I had show this previously for the interactome of S. cerevisiae [2] and here I extend the analysis to show that the same is also observed for the interactome of H. sapiens. Importantly the observation does not depend on the experimental method used since removing the yeast-two-hybrid interactions does not alter the result.
Methods and Results
Protein-protein interactions for H.sapiens were obtained from the Human Protein Reference database and from two high-throughput studies excluding any interactions derived from protein complexes. I considered only proteins that were represented in this interactome (i.e. with one or more interactions).
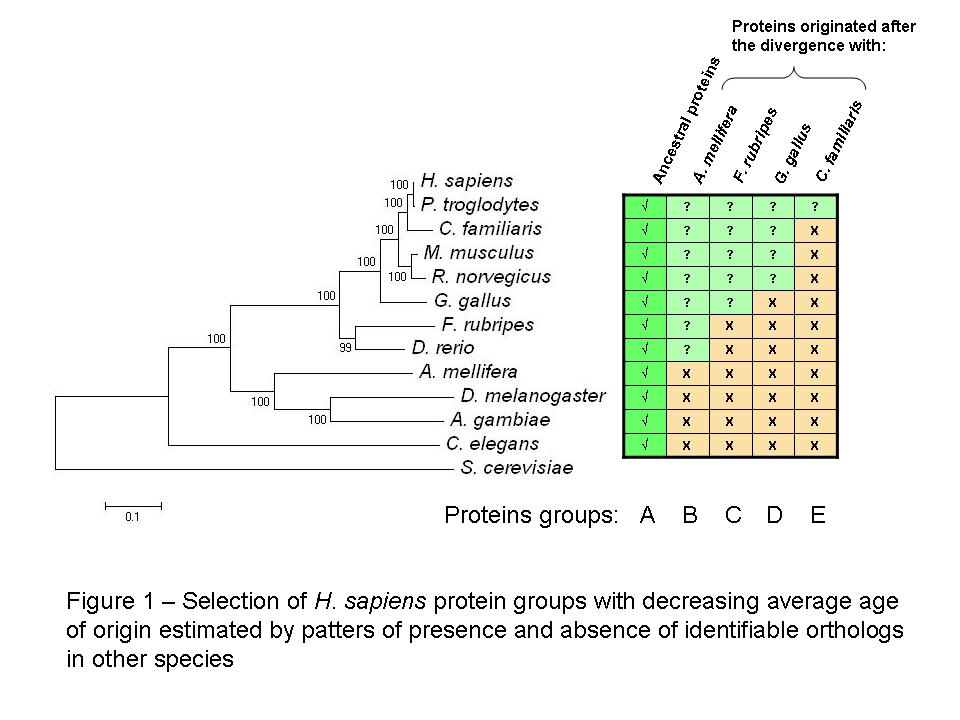
As before I created groups of H. sapiens proteins with different average age using the reciprocal best blast hit method to determine the most likely ortholog in eleven other eukaryotic species (see figure 1 for species names). For a more detailed description of the group selection and the construction of the phylogenetic tree please see the previous post [2].
It is important to note that the placement of C. familiaris does not correspond with other published phylogenetic trees it might be due to the proteins selected for the tree construction. I should consider using different combinations of ancestral proteins to check the robustness of the tree.
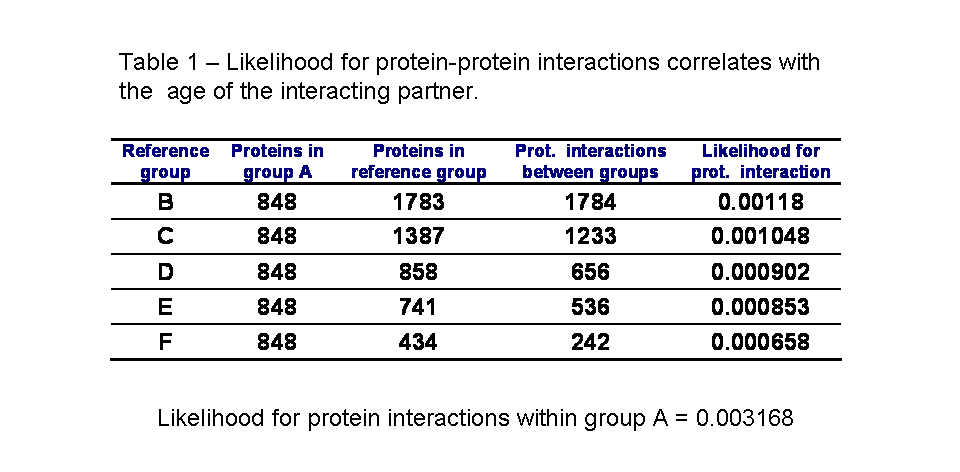
In table 1 we can see the likelihood for protein interactions to occur within the ancestral proteins of group A and between the ancestral proteins and other groups of decreasing average age. As published by Qin et al. and as I had observed before for S. cerevisiae, the interactions within groups of the same age (group A) are more likely than between groups of proteins of different times of origin. Also, the likelihood for a protein to interact with an ancestral protein depends on the age of this protein. Confirming the pervious observation that the younger the protein is the less likely it is to interact with an ancestral protein.

I redid the analysis excluding yeast-two-hybrid interactions from the dataset. As it can be see in table 2, the results are qualitatively the same. There is a small increase in the likelihood of interaction with the ancestral proteins for the youngest group (highlighted in red in table 2) that is likely due to lack of data.

Caveats and possible continuations
I still have to test the statistical significance of these observations and control for possible other effects like protein size and protein expression that could explain these results.
I am interested in continuing this further as an open project. Fallowing the suggestion of Roland Krause I will soon start a wiki page to dump the data bits accumulated for open discussion. Hopefully more people will join in and maybe we can together shape up a small communication.
[1]Qin H, Lu HH, Wu WB, Li WH. Evolution of the yeast protein interaction network. Proc Natl Acad Sci U S A. 2003 Oct 28;100(22):12820-4. Epub 2003 Oct 13
[2]Beltrao. P The likelihood that two proteins interact might depend on the proteins' age Blog post
Tags: