Cellular consequences of genetic variation
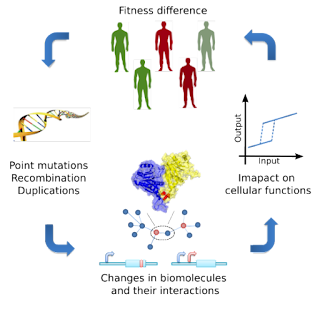
Discovering how genetic variation leads to changes in traits underlies the study of human disease and the understanding of the evolutionary process. What makes each human individual different and the diversity of life on earth are, to some extent, determined by differences in DNA. How these DNA differences manifest in a variety of shapes, capabilities and even different propensity to disease remains a mystery. Our long term goal is to have comprehensive models of how DNA changes result in differences in biomolecules, their interactions and ultimately different traits or disease.
Within this broad scope we have two major lines of research:
Trait variation among individuals of the some species
We are broadly interested in studying the genetics of trait variation from DNA to phenotype and including the genetics of human disease. For this purpose we are developing a general purpose framework to predict the molecular consequences of DNA changes (www.mutfunc.com) and using these to guide genotype-phenotype associations. We have also been taking advantage of network based methods to study the biological processes that underlie trait variation and how these compare across traits. In the context of this work we are interested in many steps including the understanding of how mutations impact on proteins; the interactions networks and biology of different cell types; and the relation between biological processes and mechanisms to phenotype.
Function and evolution of post-translational regulatory circuits
Over 100,000 phosphorylation sites have been identified in human proteins but the function and the regulators of these post-translational modifications (PTMs) remains vastly understudied. In this line of research we aim to study the kinase regulatory networks at the whole cellular level. We develop computational and experimental methods to study the evolution and function of PTMs; reconstructing the kinase signalling network; the dynamics and cell decision properties of PTM systems and how they change in disease.
Trait variation among individuals of the some species
We are broadly interested in studying the genetics of trait variation from DNA to phenotype and including the genetics of human disease. For this purpose we are developing a general purpose framework to predict the molecular consequences of DNA changes (www.mutfunc.com) and using these to guide genotype-phenotype associations. We have also been taking advantage of network based methods to study the biological processes that underlie trait variation and how these compare across traits. In the context of this work we are interested in many steps including the understanding of how mutations impact on proteins; the interactions networks and biology of different cell types; and the relation between biological processes and mechanisms to phenotype.
Function and evolution of post-translational regulatory circuits
Over 100,000 phosphorylation sites have been identified in human proteins but the function and the regulators of these post-translational modifications (PTMs) remains vastly understudied. In this line of research we aim to study the kinase regulatory networks at the whole cellular level. We develop computational and experimental methods to study the evolution and function of PTMs; reconstructing the kinase signalling network; the dynamics and cell decision properties of PTM systems and how they change in disease.
The group is at ETH Zurich within the Institute of Molecular Systems Biology